Cerebellum: Folding into shape
A new computational tool provides insights into the structure of the cerebellum in mammals.
- Department of Psychiatry, University of Cambridge, United Kingdom
During development, the brains of most mammals (including humans) will fold into a unique pattern of grooves and ridges. Understanding how these wrinkles emerge can provide important insights into how the brain works. Most research in this area has focused on the structure of the cerebrum, the two large lobes that make up most of the brain. However, much less is known about the structure of the cerebellum – the ‘little brain’ that sits beneath the cerebrum.
The anatomical properties of the cerebrum – such as the number of folds it contains, its thickness and surface area – have been shown to vary between mammalian species and to influence cognitive processes in humans (Mota and Herculano-Houzel, 2015, Cachia et al., 2021). Unlike the cerebrum, which is folded in some mammals (such as humans) but flat in others (such as mice), the cerebellum always has a wrinkled appearance. The structure of the cerebellum is also more uniform across different species (including some invertebrates), and this has led to the assumption that its folds have less of an influence on cognition than the folds of the cerebrum. However, recent work suggests that the cerebellum may have a bigger role in cognition than previously thought (Buckner, 2013).
In 2020, Ken Ashwell of the University of New South Wales compared the volumes of the cerebellum and cerebrum in monotremes, marsupials and eutherian mammals (Ashwell, 2020). However, the study did not dive into the detailed anatomy of the cerebellum, as its small size and highly folded configuration are difficult to examine using conventional methods. Now, in eLife, Roberto Toro from the Institut Pasteur and colleagues – including Katja Heuer as first author – report a new approach for investigating the shape of the cerebellum in mammals (Heuer et al., 2023).
The team developed a computational method that can measure the surface area of the cerebellum, the shape of its individual folds, and the thickness of its most superficial layer (Figure 1A). The approach, which is freely available, was validated by ensuring that it could produce results that fitted with previously reported data. It was then applied to histological slices extracted from the cerebellums of 56 different mammals, including slices examined in the Ashwell study (Figure 1B).
Figure 1
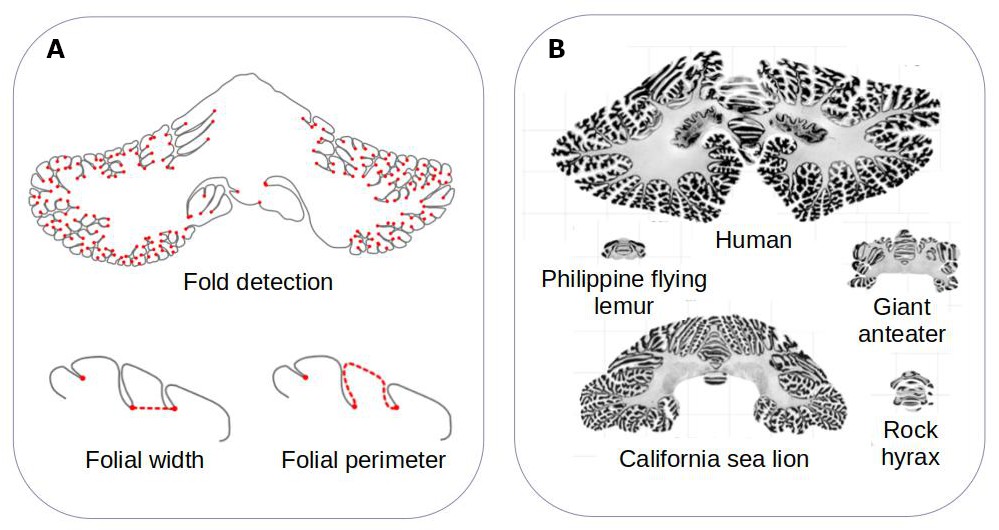
Comparing the structure of the cerebellum in mammalian species.
(A) Heuer et al. created a computational model that can automatically identify grooves in the cerebellum and measure various metrics (shown in red), including the width (bottom left) and perimeter (bottom right) of individual folds. (B) The model was applied to histological slices extracted from the cerebellums of 56 mammals. The results of this analysis are shown for five species (which are displayed on the same scale). Heuer et al. estimated that most mammals are descended from a species that had a cerebellum similar to that of the rock hyrax (bottom right).
Image credit: Adapted from Figure 1 and Figure 3 of the manuscript by Heuer et al., 2023
Heuer et al. – who are based at various institutes in France, the United Kingdom and Germany – found that the different folding metrics detected by their model could be split in to two groups: ones that varied a lot between species (such as surface area), and ones that did not vary as much (such as the width of individual folds). While body size varied by 11 orders of magnitude within the sample, the surface area of the cerebellum varied much less (by around 2.5 orders of magnitude), and the width between two folds only varied by 0.5 orders of magnitude. This suggests that larger mammals have larger cerebellums, but the size of their folds are relatively thin in comparison.
The folding metrics measured also scaled within species. For instance, bigger cerebellums had a higher number of folds than smaller ones – an effect which has also been observed in the cerebrum (Germanaud et al., 2012). Species with larger cerebrums also appeared to have relatively smaller cerebellums.
Heuer et al. then set out to find which computational model of evolution was able to recreate the anatomies of the cerebellums found in the different mammalian species. They found that a stabilising model – that is, when animals that are straying away from their common ancestor randomly evolve back to these initial properties – worked best. Using this model, Heuer et al. estimated that the organism most mammals descended from probably had a cerebellum similar to the one found in an animal known as the rock hyrax (Figure 1B).
These observations, along with other results reported by Heuer et al., provide food for thought about the mechanics of brain folding. Although the composition of cells within the cerebellum differ from those in the cerebrum, Heuer et al. propose that these parts of the brain fold in the same way, with the most superficial layer expanding more than the layer immediately below. This process, along with neurons in the cerebellum developing in a certain way, may lead to specific patterns of grooves and ridges forming in the cerebellums of different species. In the future, the new tool created by Heuer et al. could be used to see if these observations occur in the cerebellums of other species (including non-mammals), and to look in to the ‘folds within folds’ that appear in larger cerebellums in more detail.
References
Article and author information
Author details
Héloïse de Vareilles

Publication history
- Version of Record published: November 9, 2023 (version 1)
Copyright
© 2023, de Vareilles
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 756
- views
-
- 47
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Cerebellum: Folding into shape
eLife 12:e93122.
https://doi.org/10.7554/eLife.93122
Further reading
-
- Biochemistry and Chemical Biology
- Evolutionary Biology
Stramenopiles form a clade of diverse eukaryotic organisms, including multicellular algae, the fish and plant pathogenic oomycetes, such as the potato blight Phytophthora, and the human intestinal protozoan Blastocystis. In most eukaryotes, glycolysis is a strictly cytosolic metabolic pathway that converts glucose to pyruvate, resulting in the production of NADH and ATP (Adenosine triphosphate). In contrast, stramenopiles have a branched glycolysis in which the enzymes of the pay-off phase are located in both the cytosol and the mitochondrial matrix. Here, we identify a mitochondrial carrier in Blastocystis that can transport glycolytic intermediates, such as dihydroxyacetone phosphate and glyceraldehyde-3-phosphate, across the mitochondrial inner membrane, linking the cytosolic and mitochondrial branches of glycolysis. Comparative analyses with the phylogenetically related human mitochondrial oxoglutarate carrier (SLC25A11) and dicarboxylate carrier (SLC25A10) show that the glycolytic intermediate carrier has lost its ability to transport the canonical substrates malate and oxoglutarate. Blastocystis lacks several key components of oxidative phosphorylation required for the generation of mitochondrial ATP, such as complexes III and IV, ATP synthase, and ADP/ATP carriers. The presence of the glycolytic pay-off phase in the mitochondrial matrix generates ATP, which powers energy-requiring processes, such as macromolecular synthesis, as well as NADH, used by mitochondrial complex I to generate a proton motive force to drive the import of proteins and molecules. Given its unique substrate specificity and central role in carbon and energy metabolism, the carrier for glycolytic intermediates identified here represents a specific drug and pesticide target against stramenopile pathogens, which are of great economic importance.
-
- Evolutionary Biology
- Genetics and Genomics
A protein’s genetic architecture – the set of causal rules by which its sequence produces its functions – also determines its possible evolutionary trajectories. Prior research has proposed that the genetic architecture of proteins is very complex, with pervasive epistatic interactions that constrain evolution and make function difficult to predict from sequence. Most of this work has analyzed only the direct paths between two proteins of interest – excluding the vast majority of possible genotypes and evolutionary trajectories – and has considered only a single protein function, leaving unaddressed the genetic architecture of functional specificity and its impact on the evolution of new functions. Here, we develop a new method based on ordinal logistic regression to directly characterize the global genetic determinants of multiple protein functions from 20-state combinatorial deep mutational scanning (DMS) experiments. We use it to dissect the genetic architecture and evolution of a transcription factor’s specificity for DNA, using data from a combinatorial DMS of an ancient steroid hormone receptor’s capacity to activate transcription from two biologically relevant DNA elements. We show that the genetic architecture of DNA recognition consists of a dense set of main and pairwise effects that involve virtually every possible amino acid state in the protein-DNA interface, but higher-order epistasis plays only a tiny role. Pairwise interactions enlarge the set of functional sequences and are the primary determinants of specificity for different DNA elements. They also massively expand the number of opportunities for single-residue mutations to switch specificity from one DNA target to another. By bringing variants with different functions close together in sequence space, pairwise epistasis therefore facilitates rather than constrains the evolution of new functions.




{kind=link}